a metaphor-heavy intro to de novo peptide sequencing
2017-08-11; tl;dr: explanation of protein identification with mass spectrometry de novo (without a database)
Proteins are the key functional components of all cells (i.e. the things that do things). To identify the amino acid sequence of proteins, the dominant player in the arena is Tandem Mass Spectrometry (aka MS/MS). MS/MS can break things up, and measure the broken parts weights’. Identifying the sequence from a series of weight measurements (more accurately, mass to charge, m/z) is similar to the following problem:
You have a watch (a protein), you smash it with a hammer (fragment the protein), and end up with a smattering of parts. It happens that you are only able to weigh the parts, ending up with a list of weights. From these weights, you have to figure out what brand and model the watch was. (Analogy from [1])
More accurately though, we typically examine peptides (chunk of a protein) rather than proteins, because peptides run through liquid chromatography more consistently and are easier to ionize (among other practical reasons). Every time you fragment a peptide, you end up with a “MS/MS” spectrum - you can think of it as a peptide signature. So what do we do with this peptide signature?
One of two things - we can look for similar signatures in a database (aka “database search” or “spectral library search”) and infer the peptide from how similar the signature is (ie with a scoring function). Another approach is to infer the peptide sequence from the signature (aka mass spectra) itself! Below is a schematic of the de novo sequencing problem:
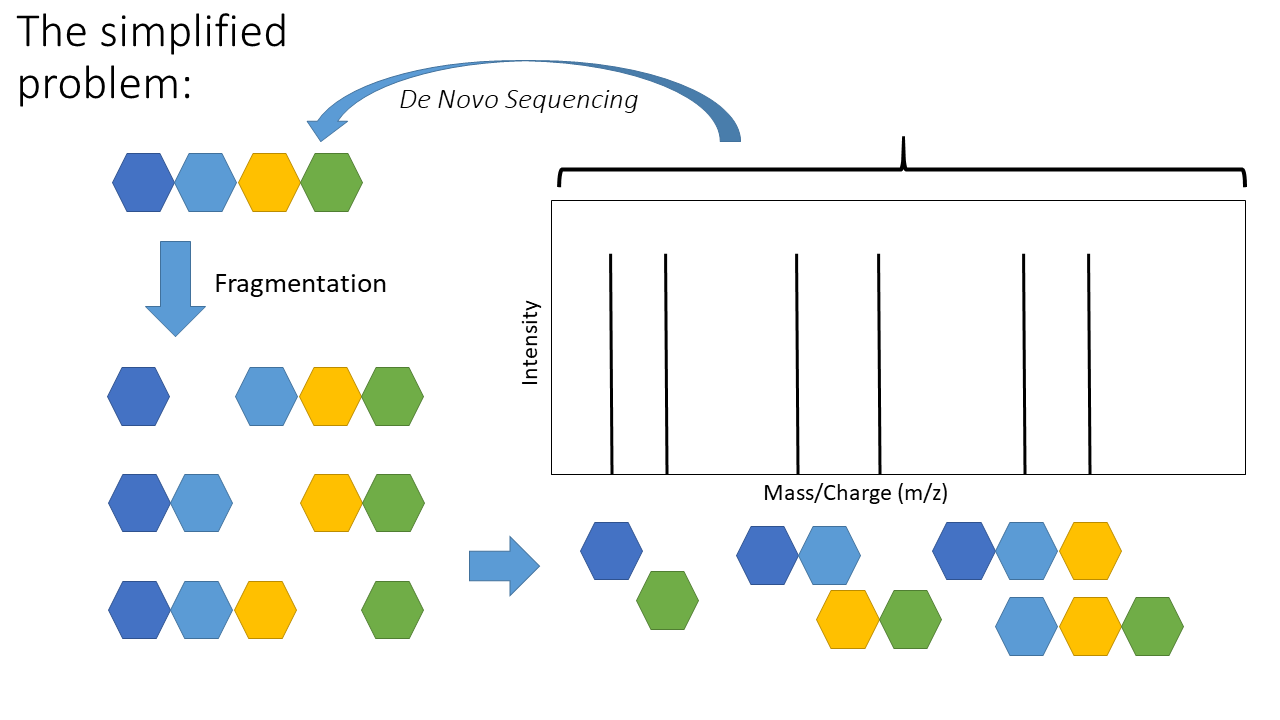
The top arrow, going from the mass spectra to the sequence, represents de novo sequencing. The key aspect of de novo sequencing is that the distance between peaks on the mass spectra are informative - they tell you about the mass difference between fragments. Those mass differences match the mass of an amino acid! See schematic below demonstrating this.
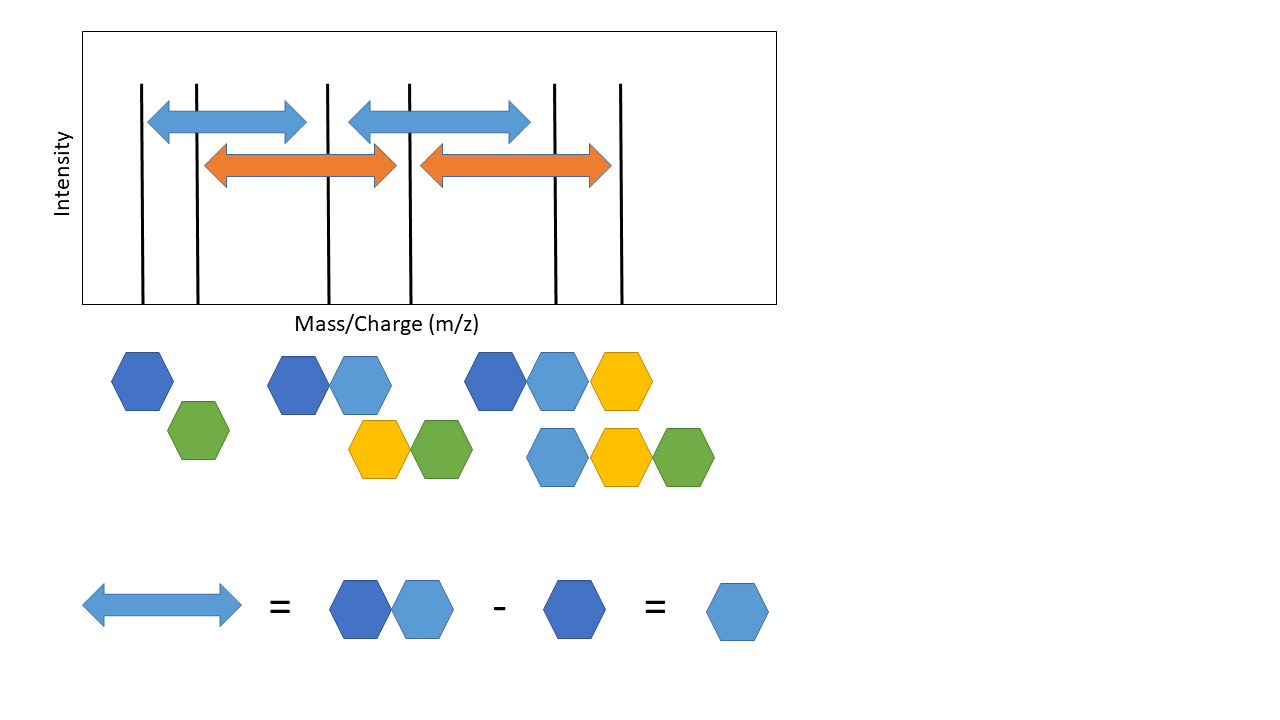
This is the basic premise of de novo peptide sequencing. There are many different approaches to solving this problem computationally. Interestingly, de novo sequencing is more similar to database search that is sometimes presented. The key difference is that a database is generated from scratch in de novo sequencing (this isn’t true for every de novo approach, but most). Also, I’ve simplified a few of the practical challenges. Notice how the arrows above are colour coded - not all distances between each peak represents the mass of an amino acid! There is also another key issue - other components of the peptide can also be knocked off during fragmentation (“neutral losses”). This can make the mass spectra noisy with extra peaks, although some algorithms use this information to increase confidence in the scoring of specific amino acids (e.g. PepNovo [2]).
PepNovo is a great example of a de novo approach. The paper is quite technical, but I highly recommend spending some time with it. It’s a very cool demonstration of the power of Probabilistic Graphical Models. I was originally going to try and summarize it in this post, but maybe I’ll leave that to another time - we’ll see if anybody ever reads this!
[1] Marcotte, E. M. 2007. How do shotgun proteomics algorithms identify proteins? Nature biotechnology, 25: 755–757.
[2] Frank, A., and Pevzner, P. 2005. PepNovo: De Novo Peptide Sequencing via Probabilistic Network Modeling. Analytical Chemistry, 77: 964–973.